Carbocations: Properties, Formation, and Stability
- Carbocations are electron-deficient species with an empty p-orbital
- Lacking a full octet and bearing a positive charge, they tend to be fairly high-energy (i.e. unstable) species and are often encountered as transient intermediates in many chemical reactions.
- Three main factors increase the stability of carbocations:
- Increasing the number of adjacent carbon atoms: methyl (least stable carbocation) < primary < secondary < tertiary (most stable carbocation)
- Adjacent pi bonds that allow the carbocation p-orbital to be part of a conjugated pi-system system (“delocalization through resonance“)
- Adjacent atoms with lone pairs that can provide the carbon with a full octet.
- Adjacent electron withdrawing groups that are unable to donate a lone pair (e.g. CF3, NO2 ) greatly decrease the stability of carbocations.
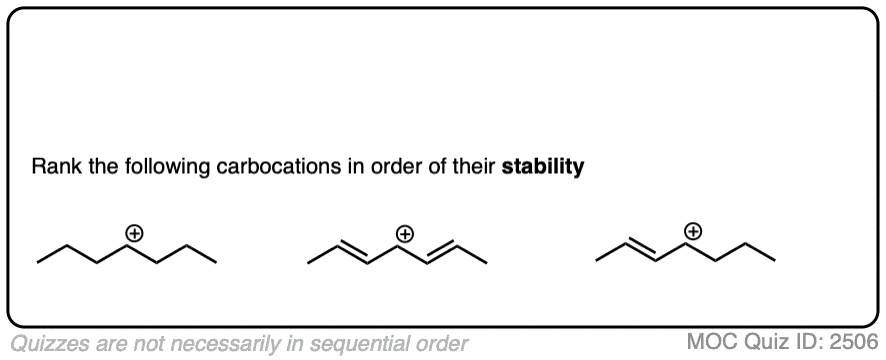
- Additionally, carbocations decrease in stability in the order alkyl (most stable) > alkenyl > alkynyl (least stable)
- A few additional factors that influence carbocation stability include aromaticity / antiaromaticity, planarity (bridgehead carbocations are unstable) and special cases involving small rings.
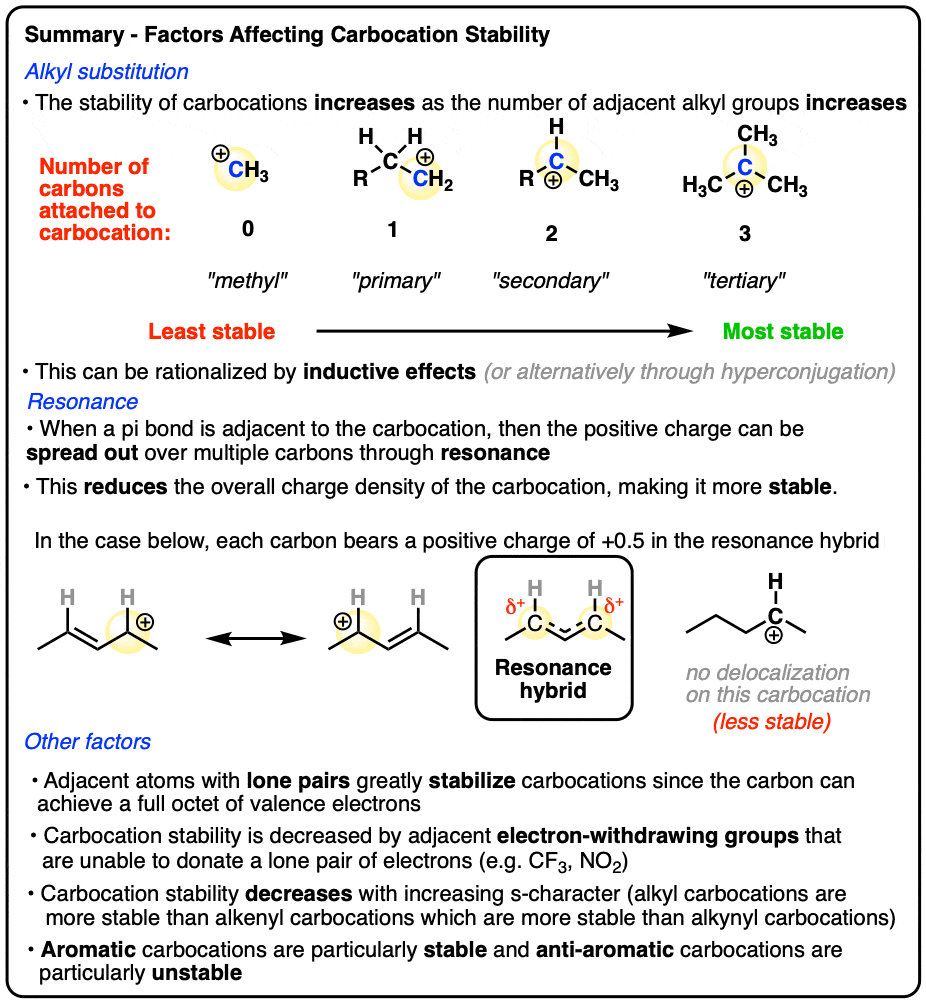
Table of Contents
- What Is A Carbocation?
- Formation of Carbocations
- Factors That Stabilize Carbocations – Substitution
- Factors That Stabilize Carbocations – Resonance
- Applying Carbocation Stability To Understand Reactions
- Stabilization By Adjacent Lone Pairs
- What’s More Important – Resonance Or Substitution?
- Inductive Effects
- Some Special Cases
- Summary
- Notes
- Quiz Yourself!
- (Advanced) References and Further Reading
1. What is A Carbocation?
A positively charged carbon atom bearing three covalent bonds and an empty orbital is called a carbocation (or more officially, a “carbenium” ion, although for our purposes we’re going to use “carbocation” [Note 1 ])
Other than the charge of +1 on the central carbon, the structure and properties of the vast majority [Note 2] of carbocations closely resemble those of neutral boron compounds.

Both carbocations and neutral boron compounds generally have
- an sp2-hybridized central atom
- with 6 valence electrons,
- an empty p-orbital,
- trigonal planar geometry
- and bond angles of 120°.
[How do we know this? X-ray crystallography Note 3]
Like boron compounds, carbocations are electron-deficient Lewis acids that will readily combine with Lewis bases, resulting in a tetrahedral, sp3-hybridized atom with a full octet of electrons.
See if you can draw the curved arrow for the reaction below.
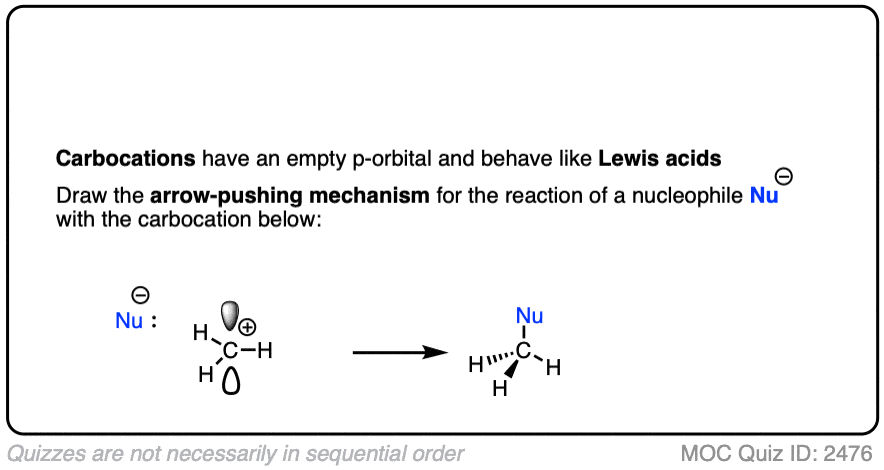 Click to Flip
Click to Flip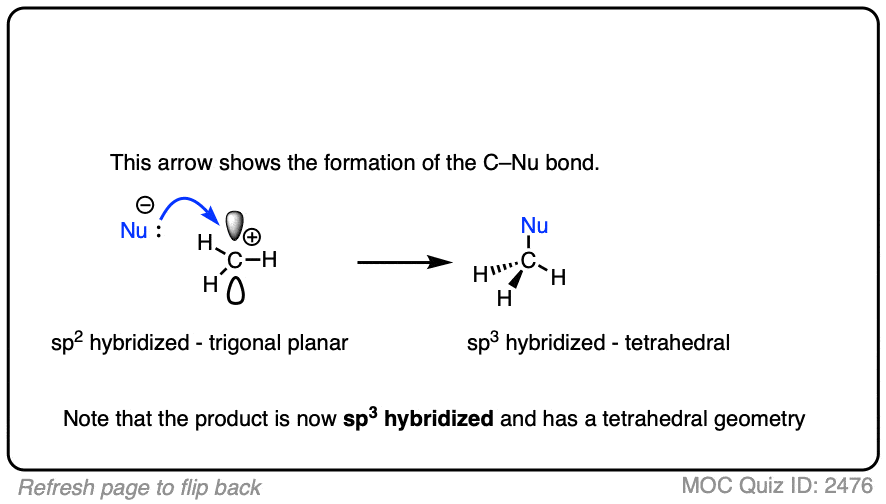
Due to their trigonal planar geometry, carbocations can undergo attack from either face of the empty p-orbital.
Be alert for situations where this can result in a pair of stereoisomers.
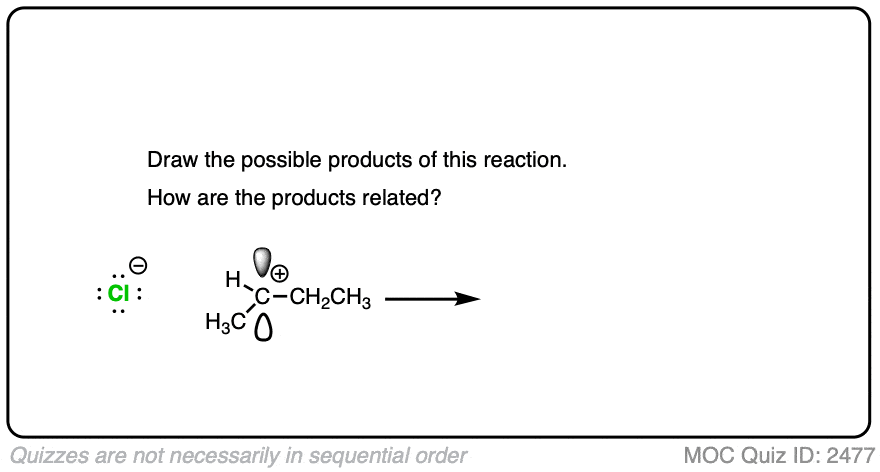 Click to Flip
Click to Flip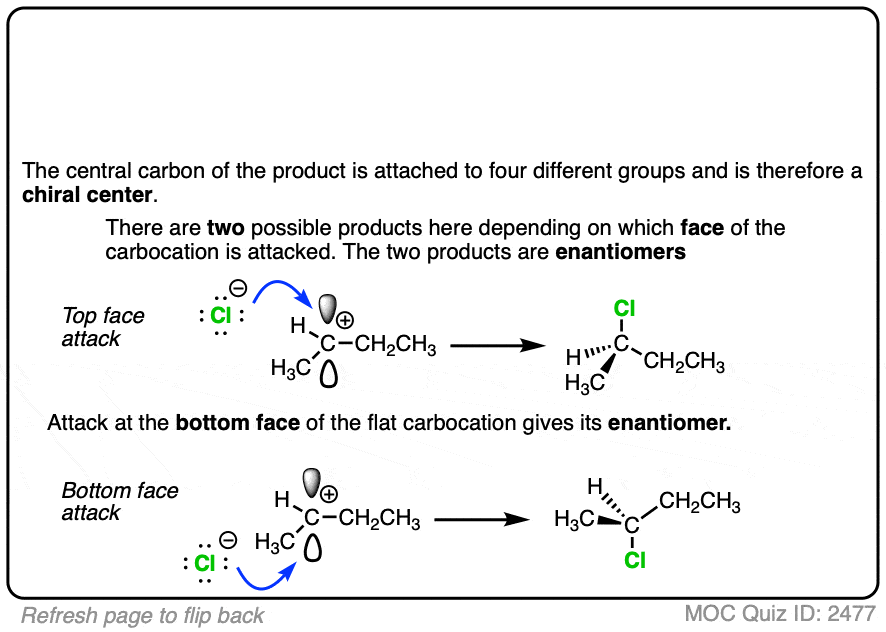
2. Formation of Carbocations
Carbocations are important intermediates in many reactions. (An intermediate, as opposed to a transition state, is a potentially isolable species in a reaction and occupies a potential energy minimum in a reaction coordinate diagram. Transition states have partial bonds, extremely short lifetimes and cannot be isolated).
Lacking a full octet of electrons around carbon and bearing a positive charge, carbocations are higher in energy and more unstable than neutral carbon compounds.
Some prominent reactions that involve carbocation intermediates are the addition of hydrogen halides (e.g. HBr, HCl, and HI) to alkenes and unimolecular substitution (SN1) and elimination (E1) reactions of alkyl halides.
In the SN1/E1 reactions, the first step is loss of a good leaving group (generally, a very weak base – See “What Makes A Good Leaving Group“) to give a carbocation. This is usually done in a highly polar solvent such as H2O or acetic acid which can help to stabilize the charged carbocation intermediate.
See if you can draw the curved arrow for formation of the carbocation from this tertiary alkyl halide, along with the transition state:
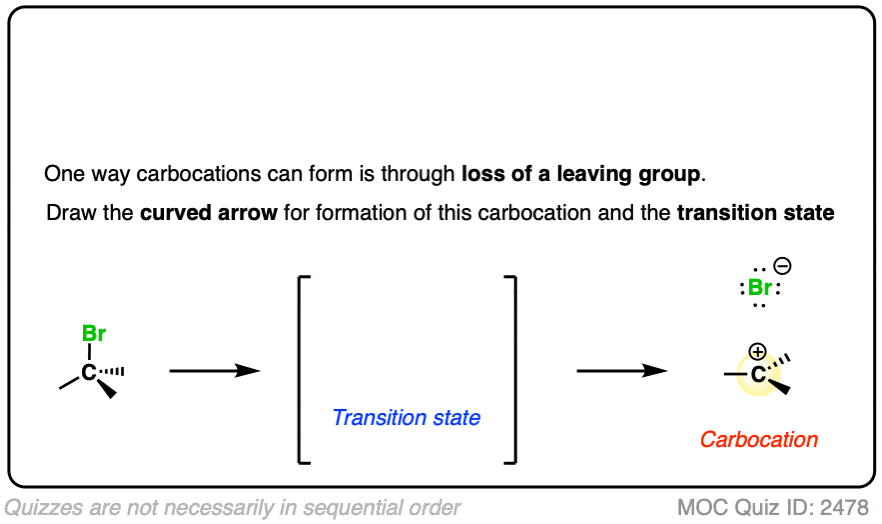 Click to Flip
Click to Flip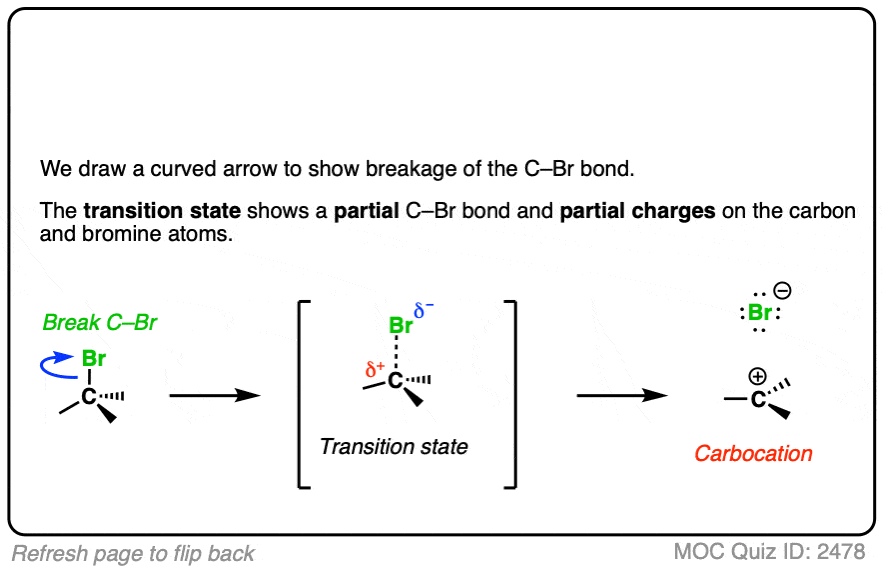
In additions of hydrohalic acids to alkenes, the first step is attack of H+ by the pair of electrons in the C-C pi bond to give a carbocation intermediate.
See if you can draw the curved-arrow pushing mechanism and the transition state for this reaction:
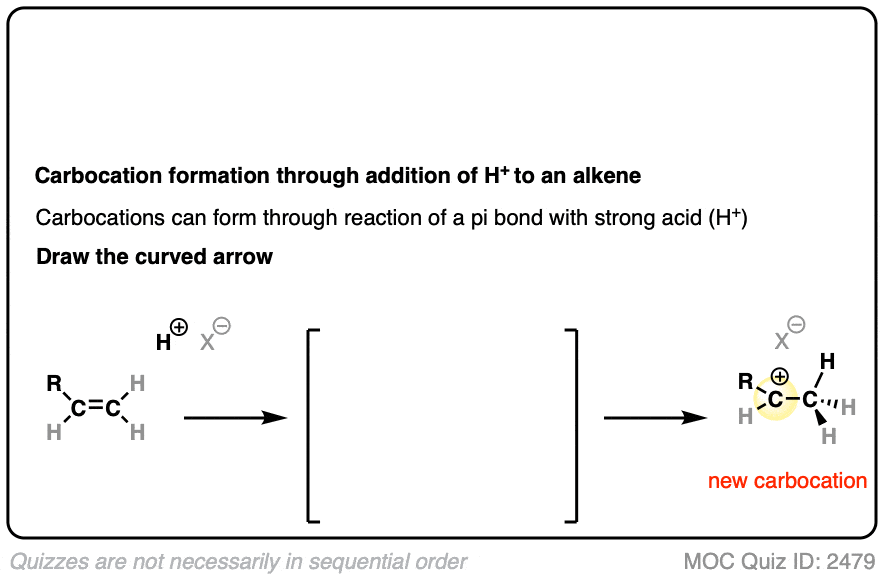 Click to Flip
Click to Flip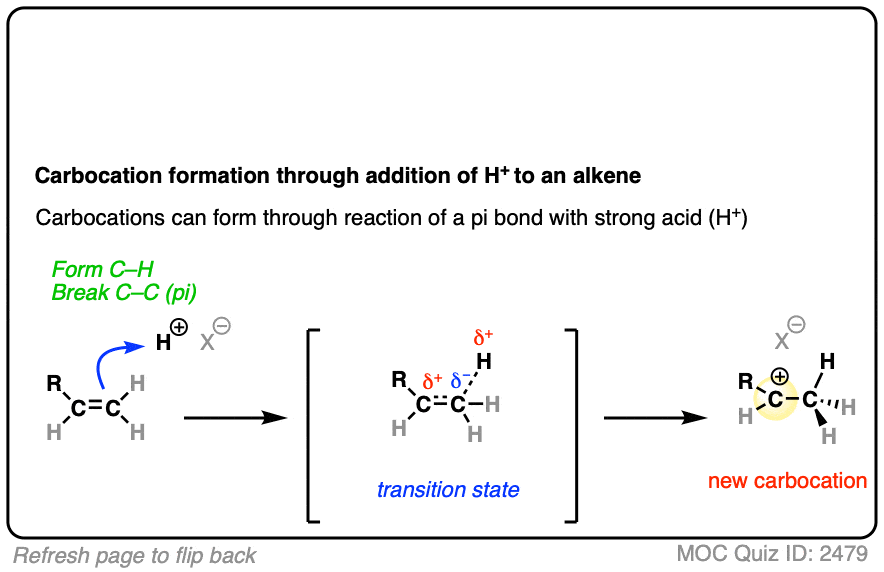
In both of these reactions, we start with a relatively low-energy starting material that passes through a high energy transition state en route to the carbocation intermediate.
In the second step, the carbocation then reacts with a Lewis base in solution to give a product where the carbon is again tetrahedral and sp3 hybridized.
A sketch of the reaction coordinates for each of these two reactions would look a little like this:
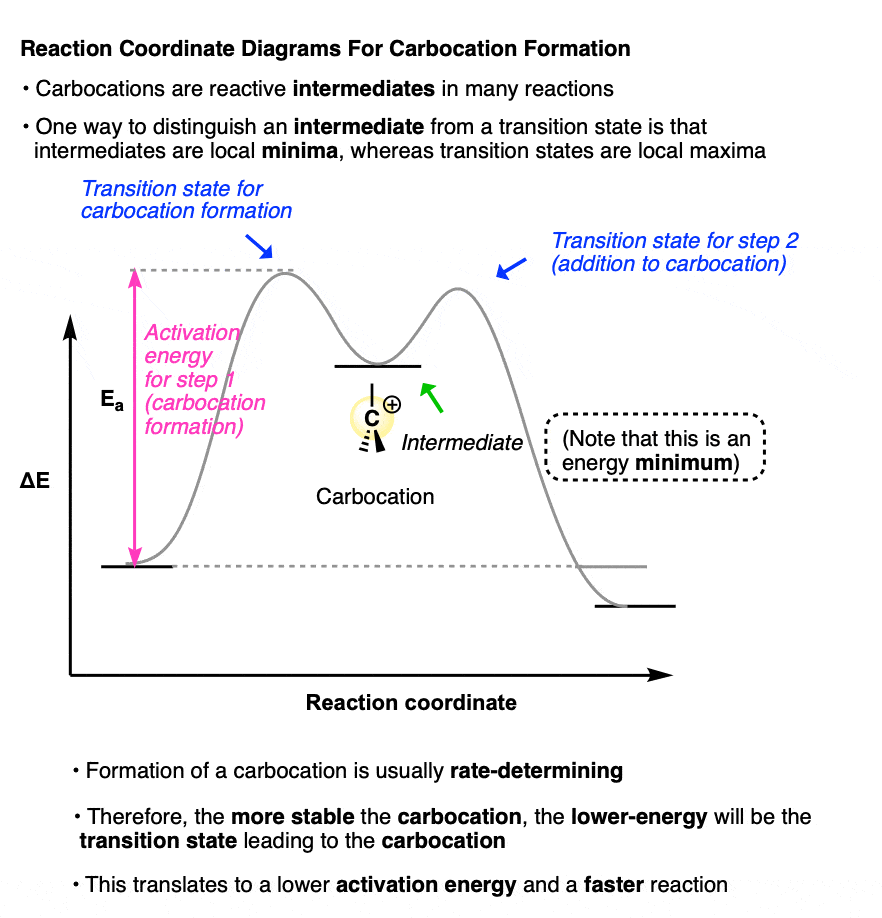
(Note that the carbocation intermediate occupies a local minimum on this reaction coordinate diagram; the two transition states are local maxima. )
Why bring this up?
Because the rate-determining step for each of these reactions is formation of the carbocation intermediate.
The more stable the carbocation intermediate, the lower in energy the transition state that leads to that carbocation, which translates into a lower activation energy and a faster reaction.
Therefore, if we understand the factors that govern the stability of carbocations, then it will also help us understand why certain reactions happen quickly whereas others do not!
3. Factors That Stabilize Carbocations – Substitution
The experimentally measured stability [Note 4] of carbocations shows the following trend:
Methyl (least stable) < primary < secondary < tertiary (most stable)
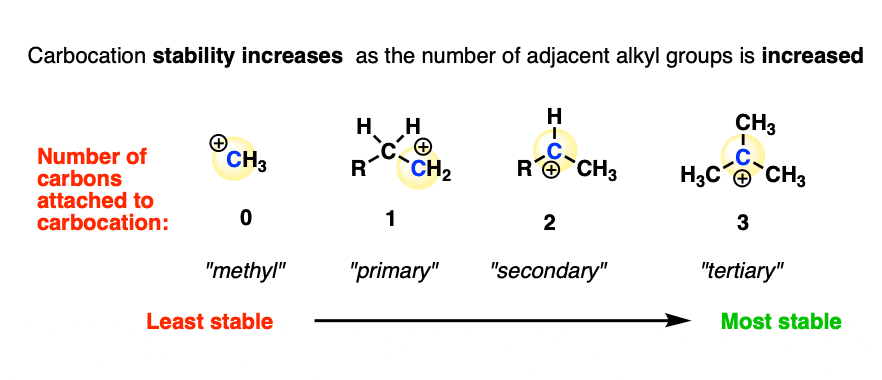
In other words, carbocation stability increases as C-H bonds are replaced with C-C bonds.
Why?
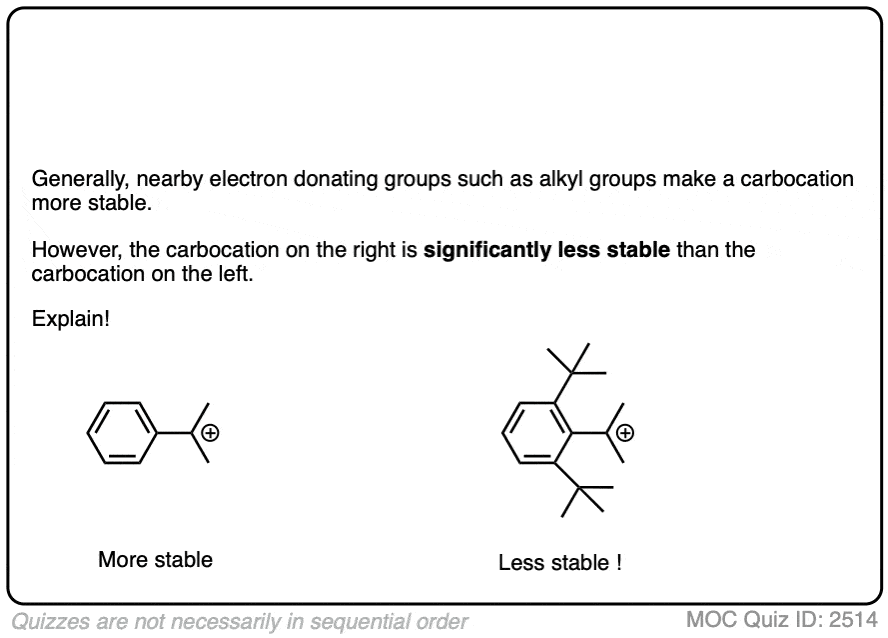
Being electron-poor, carbocations are stabilized through donation of electron density from neighboring electron-rich atoms.
As I’ve often told my students, “when you’re poor, it helps to have rich neighbors“.
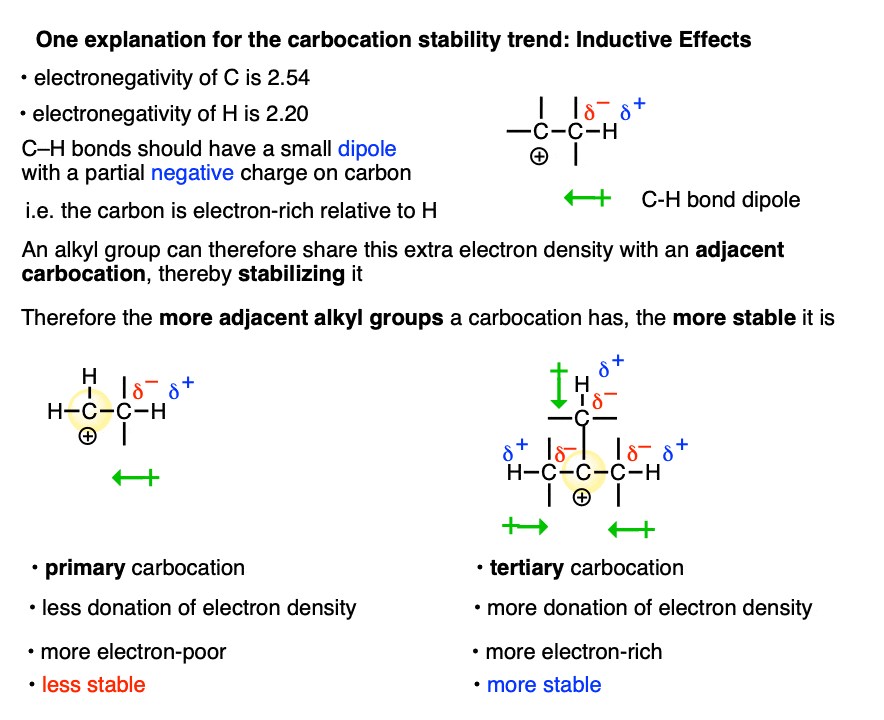
One way to rationalize this trend is through applying inductive effects.
Carbon is more electronegative (2.54) than hydrogen (2.20). So C-H bonds bear a small dipole whereby the carbon is partially negative and the hydrogen is partially positive.
The electron density from multiple C–H dipoles can add up. So the carbon of an alkyl group will have a small partial negative charge which can be donated to the adjacent carbocation, making it less electron-poor.
This isn’t possible when the carbocation is directly attached to H.
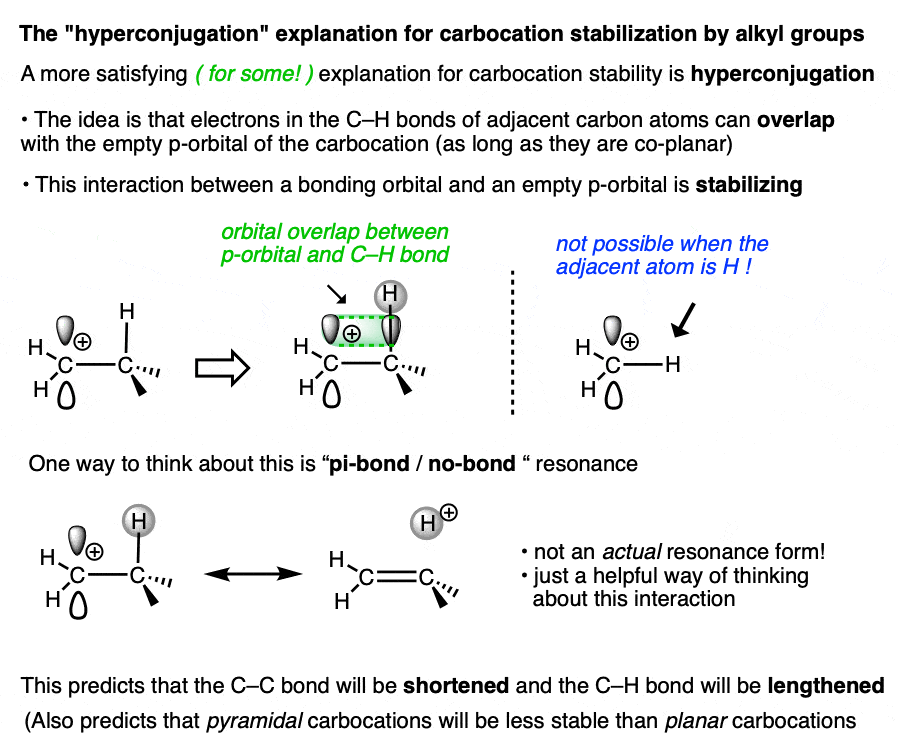
A more satisfying (to some! ) explanation comes from hyperconjugation.
Imagine lining up the two electrons in a C-H (or C-C) sigma bond with the empty p-orbital of the carbocation. Then imagine a form of “resonance” where those electrons are shared with the p-orbital to form a pi-bond (and H+).
Since electron density is being donated to an empty orbital, this should be stabilizing for the empty p-orbital (i.e. the carbocation).
We don’t have space in this article to go into all the implications of hyperconjugation in this article, but it does make some testable predictions that are borne out by experiment. [Note 3– the structure of the adamantyl carbocation is very instructive]
Whatever rationalization you prefer, the observed trend remains the same. The more substituted the carbocation, the greater its stability.
4. Factors That Stabilize Carbocations – Resonance
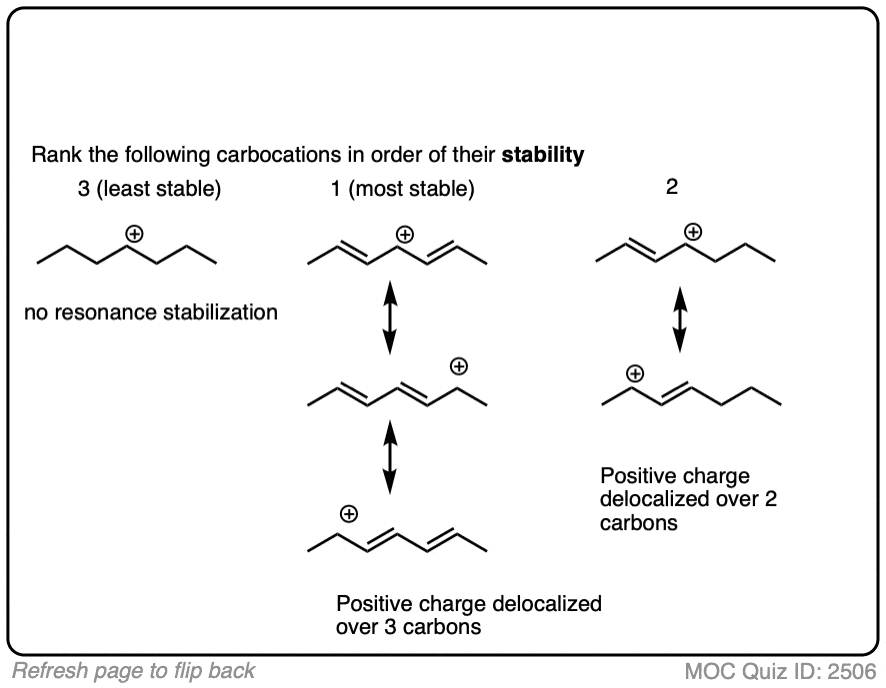
A good working principle in organic chemistry is that concentrated charge is generally more unstable (higher-energy) than dilute charge (lower-energy) [Note 5]
One place we’ve seen this before is in the concept of polarizability, where a big anion like iodide I(-) is more stable than a small ion like F(-) because the negative charge is spread out over a much greater volume. [This is the factor responsible for the greater acidity of H-I versus H-F and also the greater leaving group ability of I(-) ]
Another factor that results in this “spreading-out” of charges is “resonance”.
In a carbocation such as the n-propyl cation (CH3CH2CH2+) , the positive charge is localized to a single carbon.
But if a pi-bond is adjacent to the carbocation, we can then draw two resonance forms where the positive charge is either on C-1 or C-3.
In the resonance hybrid, the positive charge is shared between these two carbons, which will each have a charge density of +0.5 instead of +1. (This is what is meant by “resonance delocalization” – the charge has been smeared over multiple carbons)
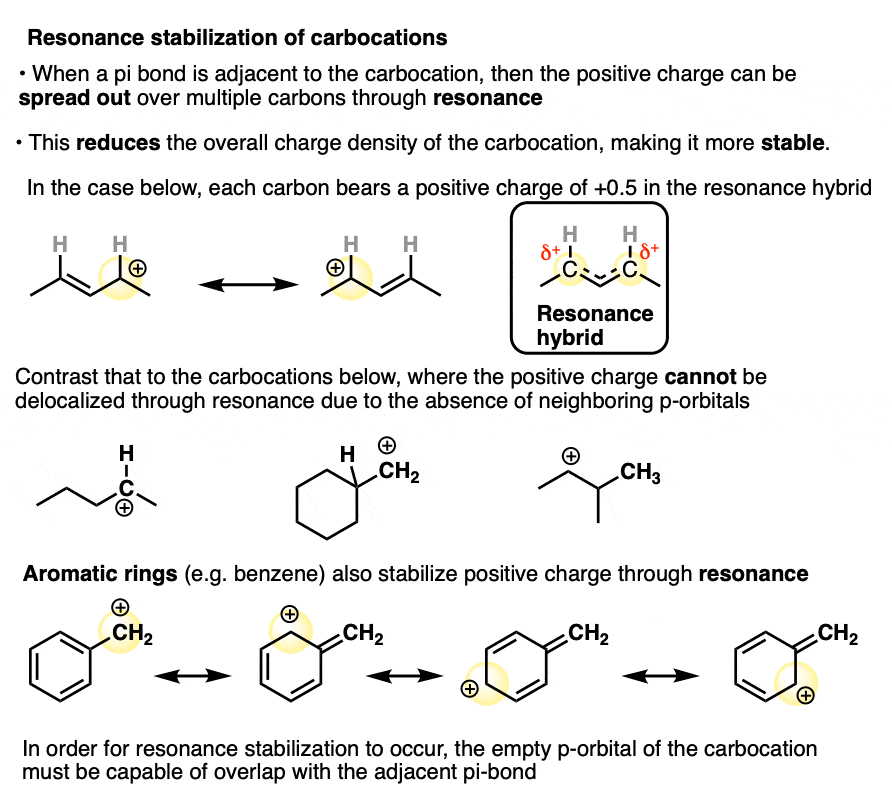
The greater stability of this carbocation (known as the allyl cation) relative to the parent propyl cation is borne out by numerous measurements. [Note 4]
A similar effect is seen when a carbocation is adjacent to an aromatic ring such as benzene. The stability of this carbocation, known as the benzyl carbocation is considerably greater than that of a “normal” primary carbocation. (In fact the benzyl carbocation is roughly as stable as the t-butyl cation, Note 4)
The stability of the carbocation tends to increase as the number of potential resonance forms increases.
For example, the “trityl” cation, Ph3C(+) is so stable that it forms a crystalline salt that can be put in a bottle and stored on a shelf indefinitely.
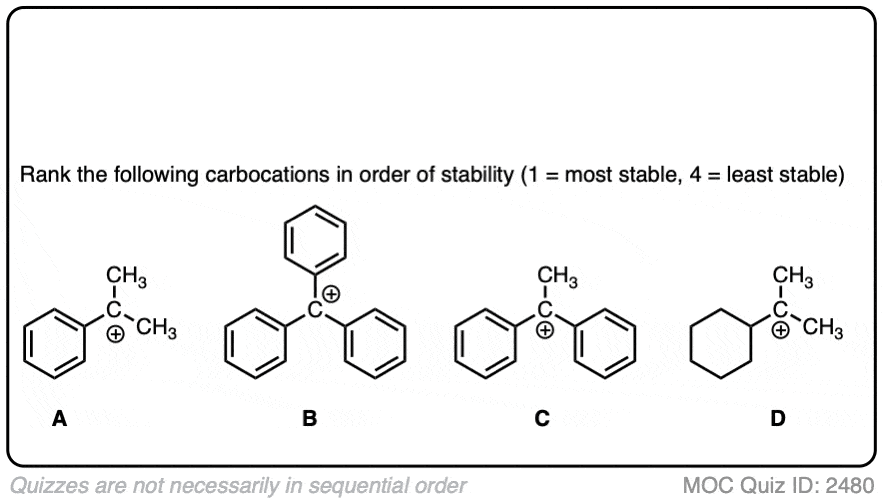 Click to Flip
Click to Flip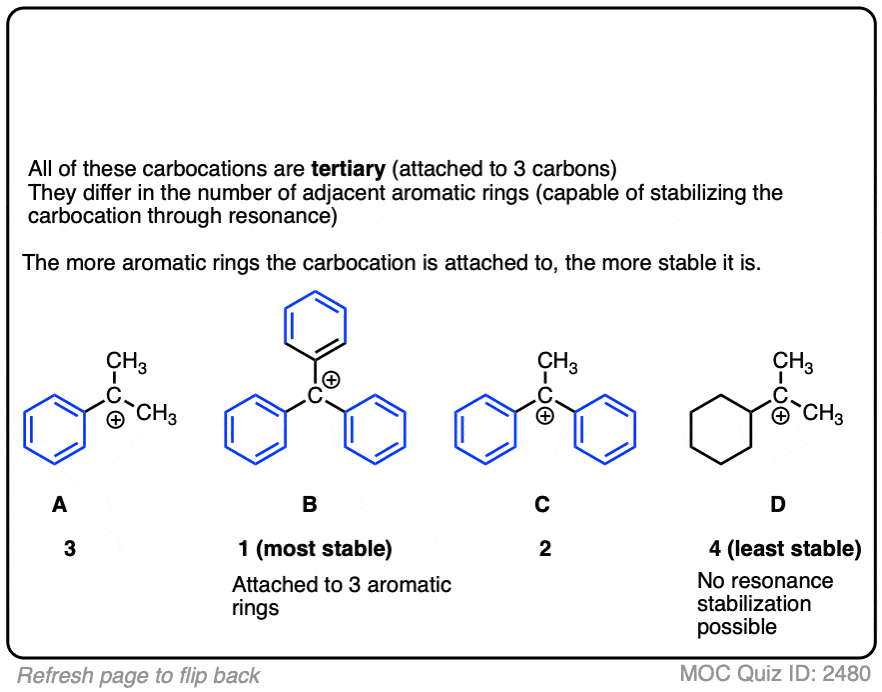
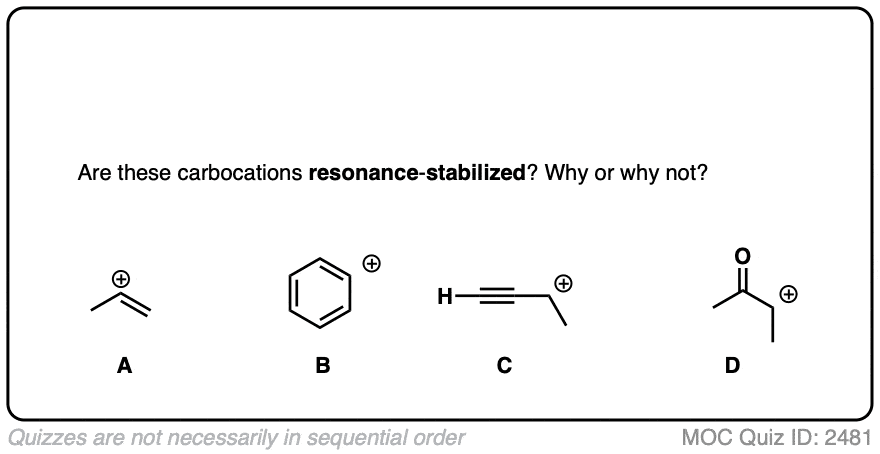
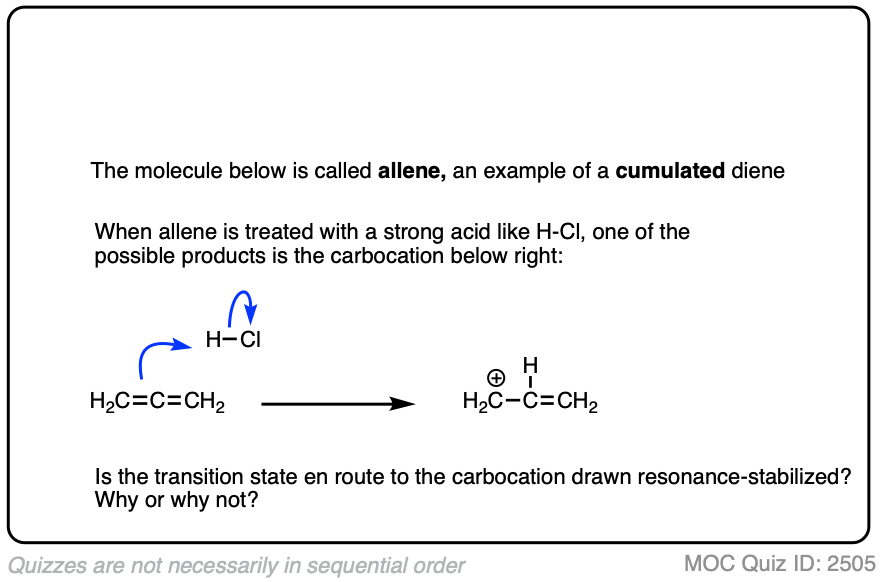
A word of warning. In order for a carbocation to be stabilized by resonance, the p-orbital of the carbocation must be able to overlap with the p-orbitals of the adjacent pi-bond. (See article – Conjugation and Resonance)
No overlap means no delocalization, which means no added stability.
Test your understanding of this concept by answering the quiz below.
 Click to Flip
Click to Flip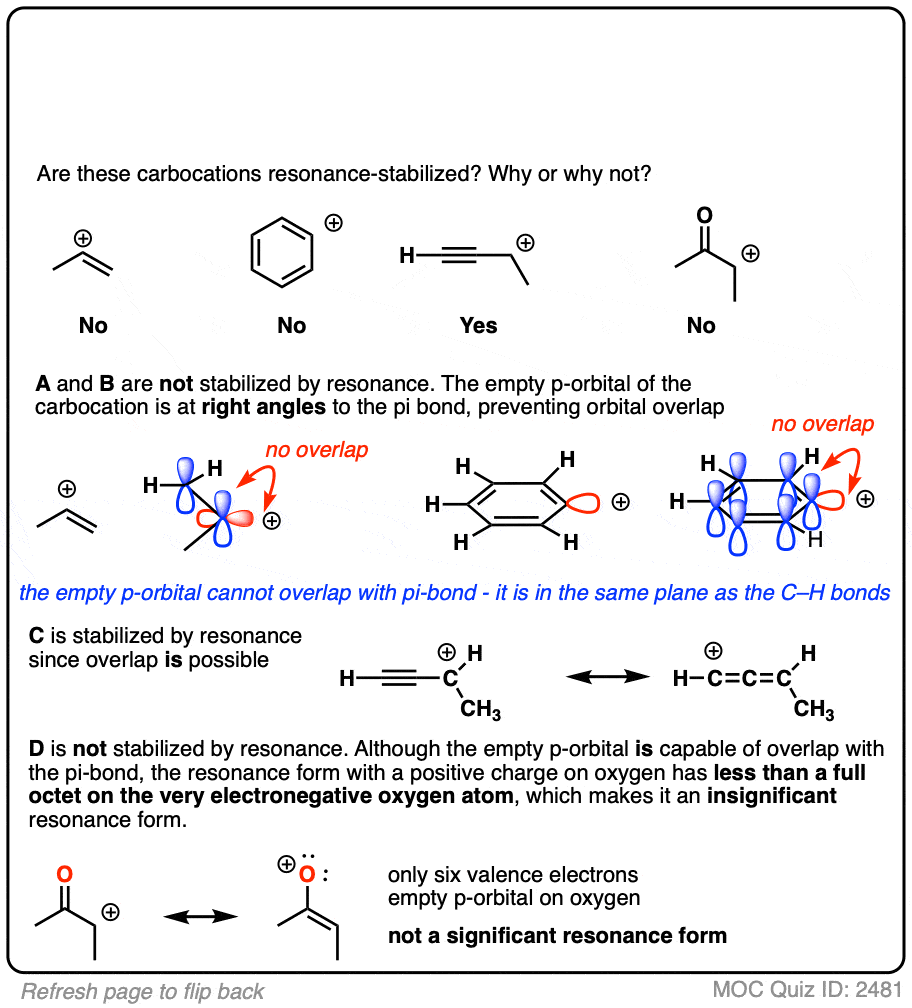
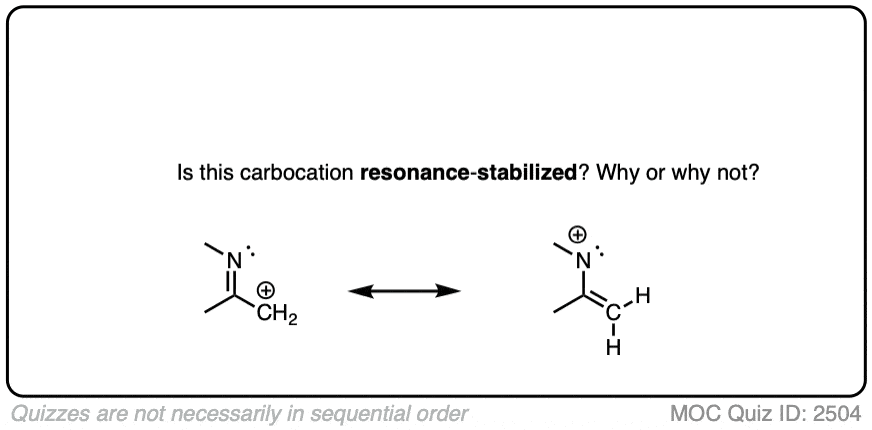
A common mistake is to think that carbocations directly attached to a pi-bond are resonance stabilized. This is not so! The pi-bond must be adjacent to the carbocation.
Another potentially counter-intuitive situation is when a carbocation is adjacent to a C=O (or C=N) bond. One might naively think that the carbocation is stabilized by resonance.
However, if you draw out the resonance form, you’ll find that in forming the C-C pi bond and breaking the C-O pi bond, you end up forming an electron-deficient species with only six valence electrons on oxygen. This “resonance form” is so high-energy as to make essentially no contribution to the resonance hybrid.
(Highly electronegative atoms like O are excellent at stabilizing negative charge, but they are terrible at stabilizing empty orbitals)
5. The Importance of Carbocation Stability In Reactions
Let’s return to the role of carbocations as intermediates in several key organic reactions.
The rate-determining step of the reaction below is protonation of the pi bond with H+ to give a carbocation intermediate.
Which of the two reactions do you think is faster?
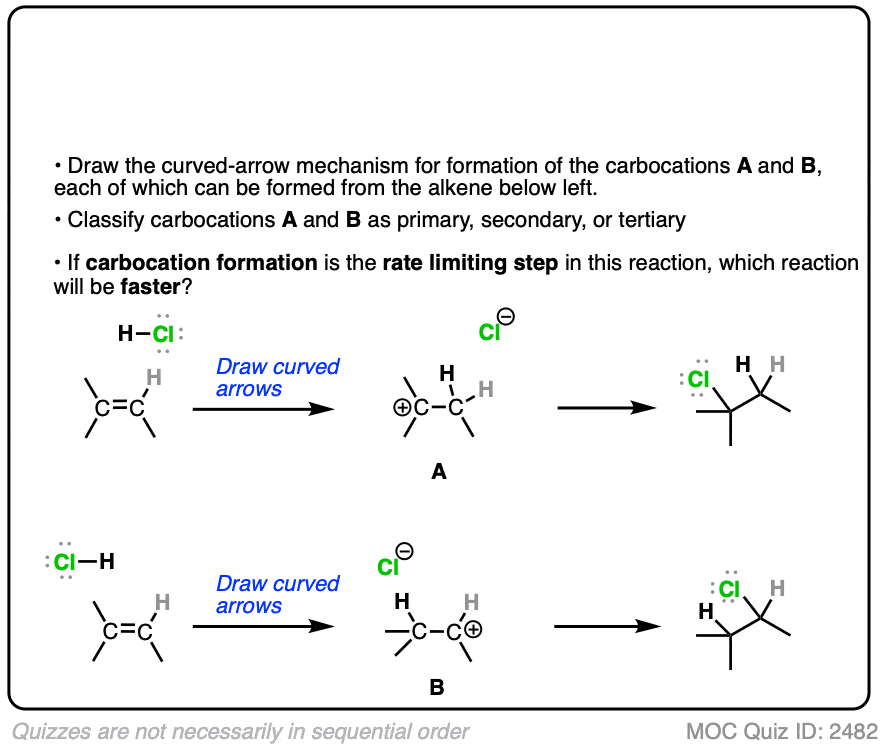 Click to Flip
Click to Flip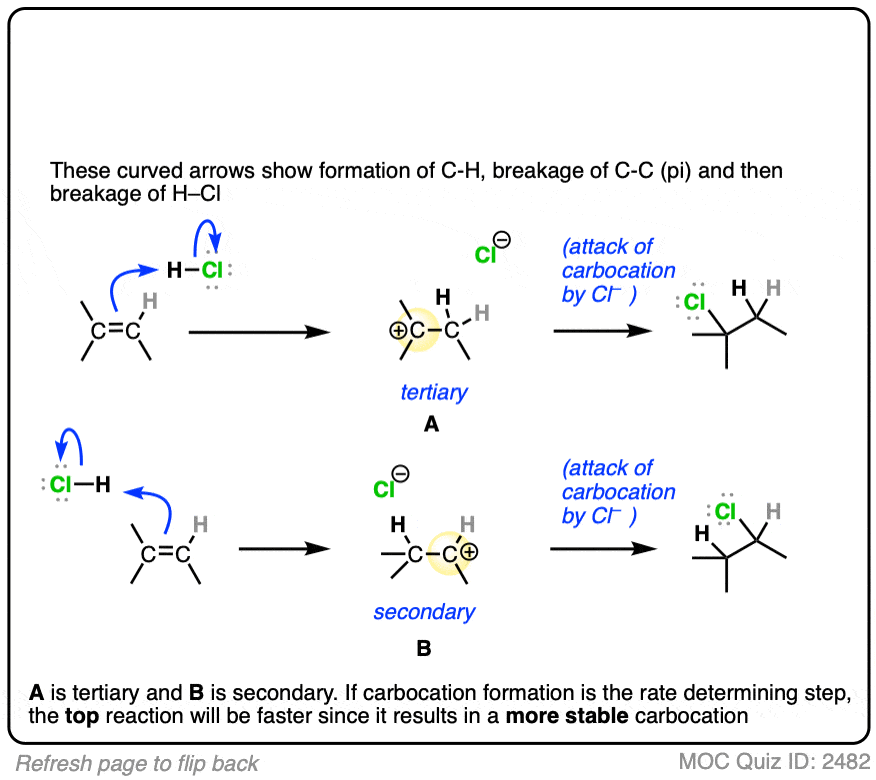
If you answered correctly, congratulations! You’ve shown you understand the reason behind Markovnikov’s rule! [See article – Markovnikov’s rule]
Let’s look at another one.
The rate-determining step of the reaction below is loss of a leaving group to form a carbocation.
Which of these two reactions proceeds faster?
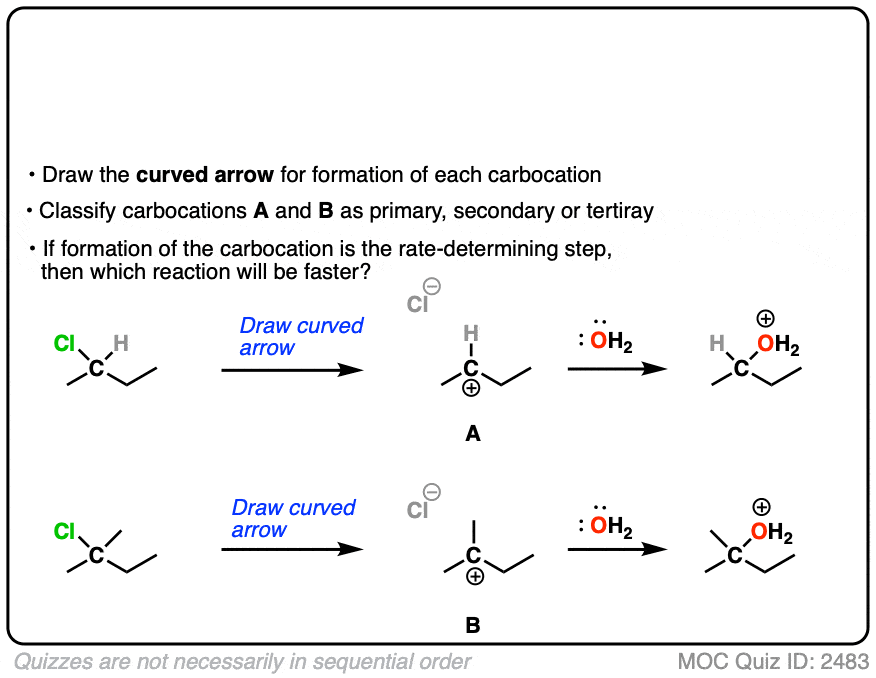 Click to Flip
Click to Flip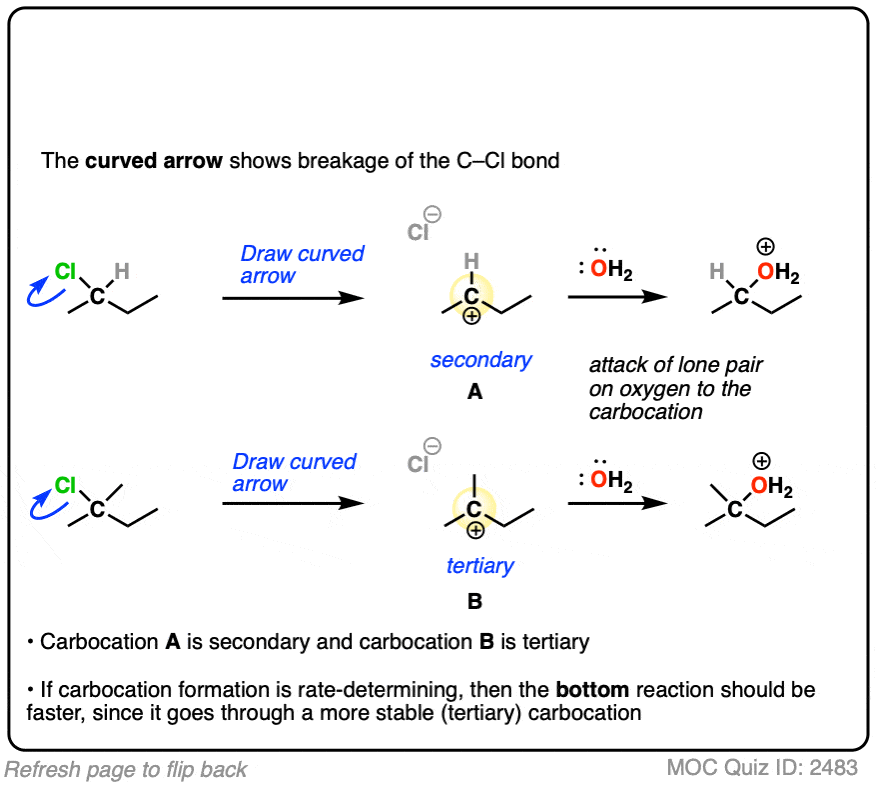
If you answered correctly, you’ve just shown an understanding one of the key factors that go into deciding whether a reaction will proceed via SN1, SN2, E1 or E2. (See article: SN1/SN2/E1/E2 – The Substrate)
6. Factors That Stabilize Carbocations – Adjacent Lone Pairs
There’s another factor that stabilizes carbocations that we haven’t touched on yet.
If a carbocation is formed adjacent to an atom bearing a lone pair (i.e. a Lewis base) then that atom can donate its pair of electrons to the carbocation, forming a new pi-bond in the process.
This is known as “pi-donation” (See article – Pi Donation)
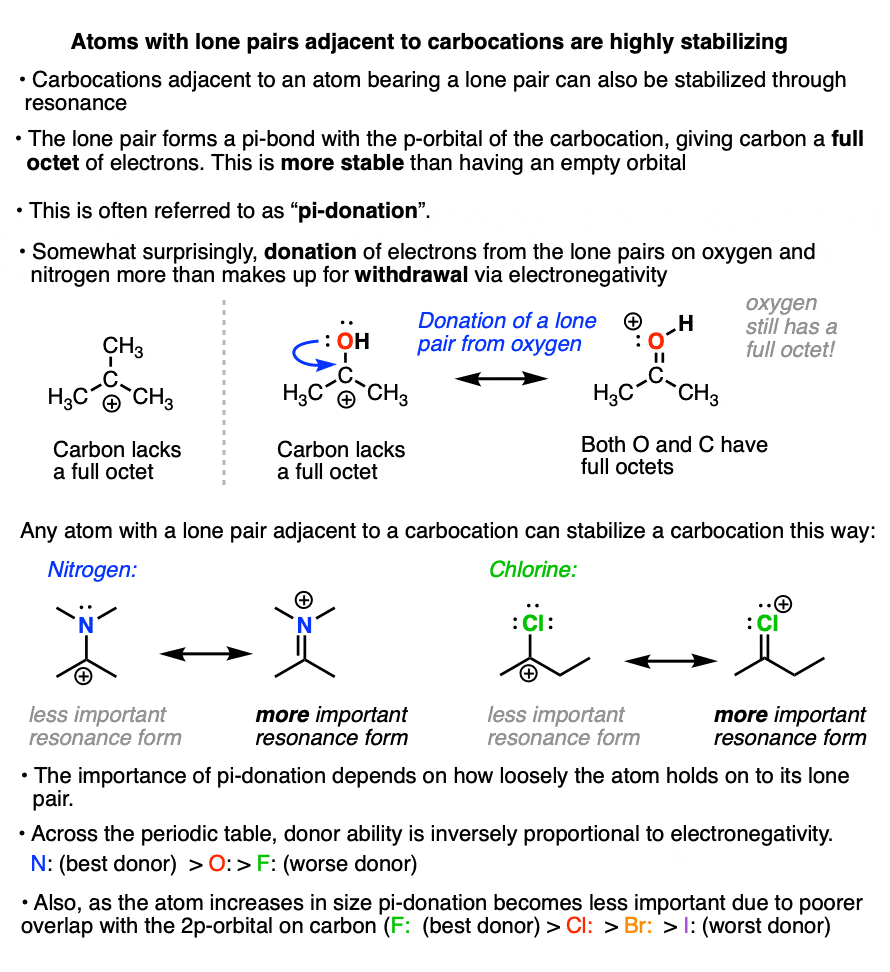
It might seem somewhat counter-intuitive that a highly electronegative atom like oxygen or nitrogen can stabilize a carbocation.
But the net result of pi-donation is that all atoms have a full octet, which is highly stabilizing. (Note that even though oxygen and nitrogen will bear a formal charge of +1, they still have a full octet of electrons! See article – How To Calculate Formal Charge)
The extent to which an atom’s lone pair can stabilize an adjacent carbocation is proportional to its basicity. The more basic the atom, the better the pi-donor.
Across a row of the periodic table, the ability to donate a pair of electrons is inversely proportional to electronegativity. So, all other factors being equal, a lone pair from nitrogen will be more stabilizing than a lone pair from oxygen, which is more stabilizing than a lone pair from fluorine.
Going down the periodic table, the ability of an atom to donate a pair of electrons is related to the ability of the atom’s valence orbital to overlap with the empty carbon 2p orbital.
Generally, as the orbital increases in size, orbital overlap (and pi-donation ability) will decrease.
Thus fluorine is a better pi-donor to carbon than chlorine, which is a better pi-donor than bromine, which is better than iodine (in iodine the valence orbitals are the 5p and 5s orbitals, which do not match up well with the smaller 2p orbital of carbon).
This is an article about carbocation stability and I want to keep it focused, but the contents of this particular section are extraordinarily important, and will come up again and again throughout organic chemistry. One application you’ll see later – Electrophilic Aromatic Substitution.
7. Which Factor is More Important – Substitution or Resonance?
So we have two factors that influence carbocation stability: substitution (primary, secondary, tertiary) and resonance.
So which is more important?
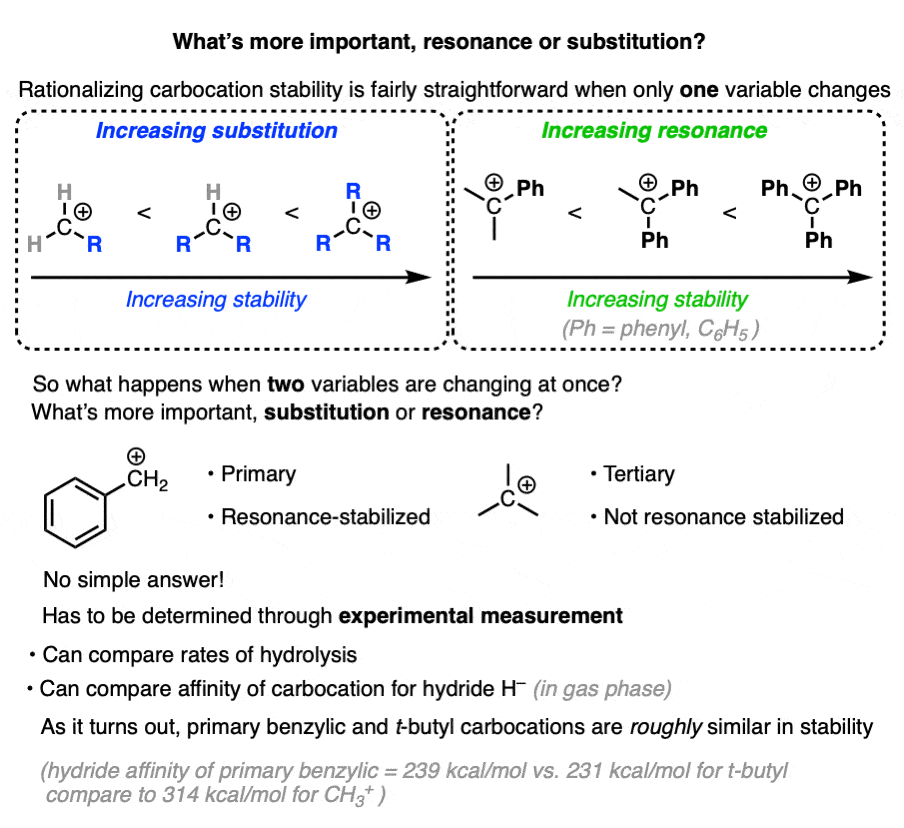
“they’re both important” is my non-answer to this question.
The actual way to answer this question is to look at experimental results.
One way to measure carbocation stability is to take a related series of alkyl halides and measure the rate of their hydrolysis in a polar protic solvent under conditions where SN1 would be likely.
(The advantage of this method is that it more closely approximates actual reaction conditions. One disadvantage is that it is really only applicable to carbocations that can form under SN1 conditions. )
Another method is to measure what is called hydride affinity – the energy released by the reaction of the carbocation with one equivalent of hydride ion (H–). (Although this can be applied to a greater range of carbocations, it is really only applicable in the gas phase).
The hydride affinity of a primary benzylic carbocation is about 239 kcal/mol versus 231 kcal/mol for a t-butyl carbocation, so these are quite comparable in energy.
A table with some values is below. See Note 4.
8. Some Factors That Destabilize Carbocations
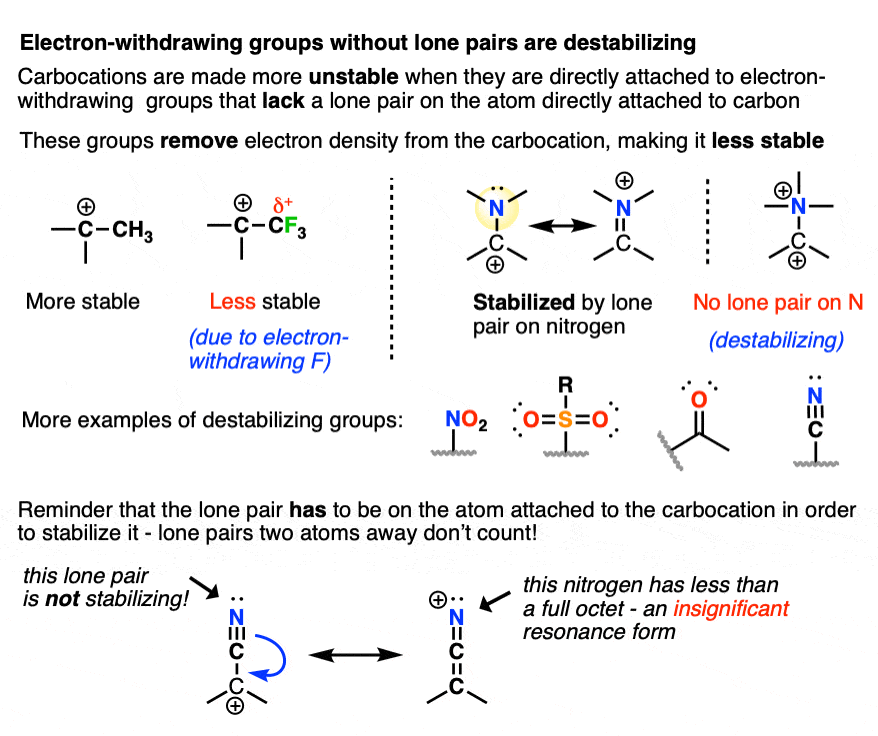
Since electron-donating groups help to stabilize carbocations, it would make sense to expect that electron-withdrawing groups will destabilize carbocations.
As we touched on above, electronegative atoms with lone pairs like O, N, and F that are directly attached to a carbocation will actually help to stabilize carbocations through pi-donation.
I would not consider those “electron-withdrawing groups” for these purposes.
By “electron-withdrawing groups” I would include
- Carbon atoms attached to electron-withdrawing groups (e.g. CF3, CCl3)
- Electronegative atoms without lone pairs (e.g. NR3(+) )
- Atoms containing a pi-bond to a more electronegative atom (e.g. C=O, CN, NO2, SO2R, etc.)
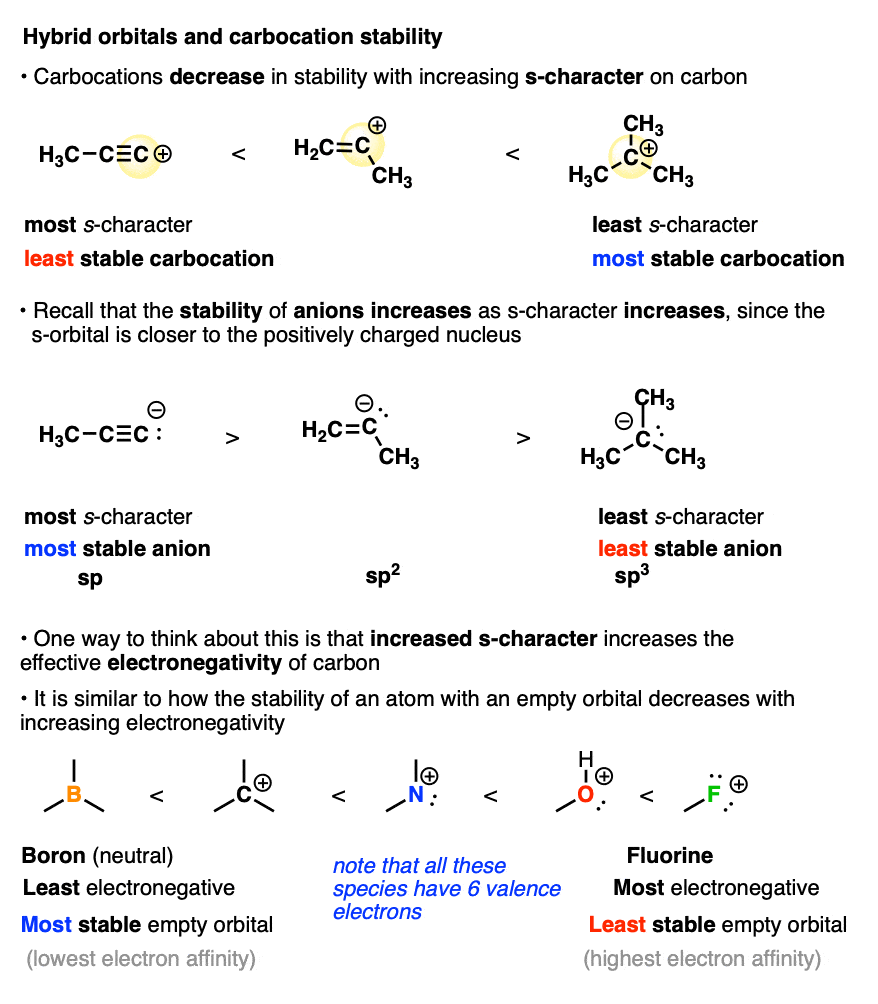
Another factor that destabilizes carbocations is the amount of s-character in the carbon atom.
You may recall that alkyne C-H bonds are particularly acidic because their sp-orbitals have 50% s-character, and since the s-orbital is closer to the positively charged nucleus, this helps to stabilize the negative charge of the conjugate base (a carbanion).
Well, when it comes to the stabilization of carbocations, this all gets thrown into reverse.
The more s-character the carbocation has, the closer that empty orbital is held to the positively charged nucleus. This has the effect of making the carbon nucleus have a greater effective electronegativity. Removing a pair of electrons to form a carbocation becomes increasingly difficult as the s-character increases. (This can be quantified by measuring hydride affinity or electron affinity)
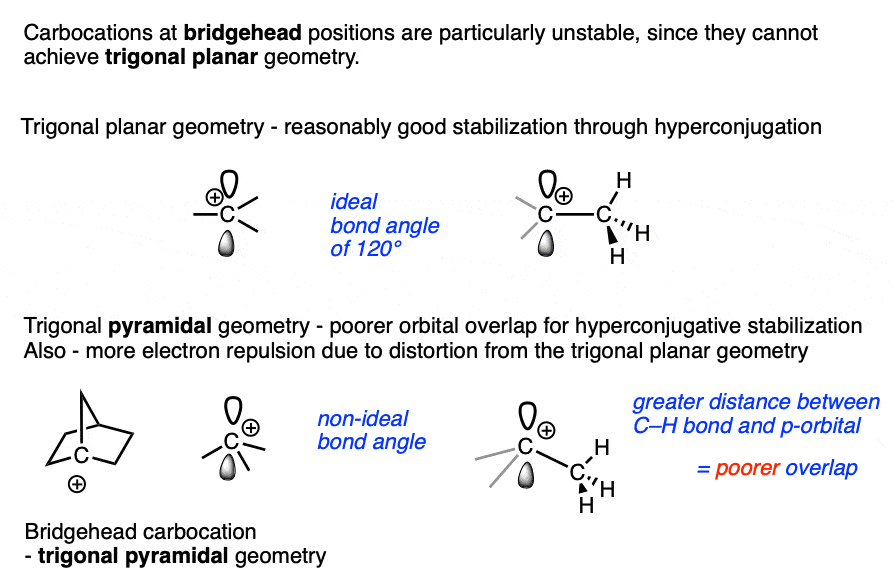
Bridgehead carbocations are also particularly unstable due to the fact that they cannot attain the ideal trigonal planar geometry, [Note 6] as are carbocations on small rings (such as the cyclopropyl cation and cyclobutyl cation). [Note 7]
9. Some Special Cases
For the sake of completeness we should probably mention one last factor that is more of a second-semester topic but has a very large impact on the stability of certain carbocations.
Some molecules have a particularly stable property known as aromaticity. (See article – Rules For Aromaticity)
A related phenomenon called antiaromaticity is responsible for the unusual instability of certain compounds. (See article – Antiaromatic Compounds and Antiaromaticity)
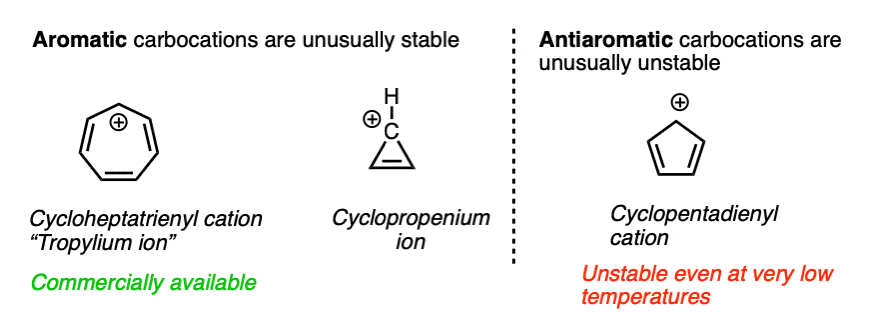
The heptatrienyl (“tropylium”) cation, C7H7+ is aromatic and due to its unusual stability, forms a stable salt that can be put in a bottle and sold commercially. The cyclopropenium ion C3H3+ is also aromatic and unusually stable.
On the other hand, the cyclopentadienyl cation, which by all appearances should be resonance-stabilized, is actually anti-aromatic and has only a fleeting existence under carefully controlled conditions at very low temperatures.
10. Summary
Carbocations are positively charged, six-electron carbon atoms with an empty p-orbital. They are important intermediates in many reactions and are highly reactive towards Lewis bases.
Three key factors stabilize carbocations:
- First, they are stabilized by adjacent alkyl groups, which can donate electron density to the electron-deficient carbon atom.
- Secondly, they can be stabilized through conjugation with pi bonds, which allows the positive charge to be delocalized through resonance.
- Third, carbocations are stabilized by atoms with a lone pair capable of forming a pi bond.
Being electron-deficient, carbocations are destabilized by strongly electron-withdrawing substituents incapable of donating electron pairs (e.g. CF3, C=O). They are also destabilized if they are unable to attain the ideal trigonal planar geometry, which can happen in bridgehead carbocations. For similar reasons, carbocations that are part of highly strained rings such as cyclopropanes and cyclobutanes tend to be quite unstable.
Notes
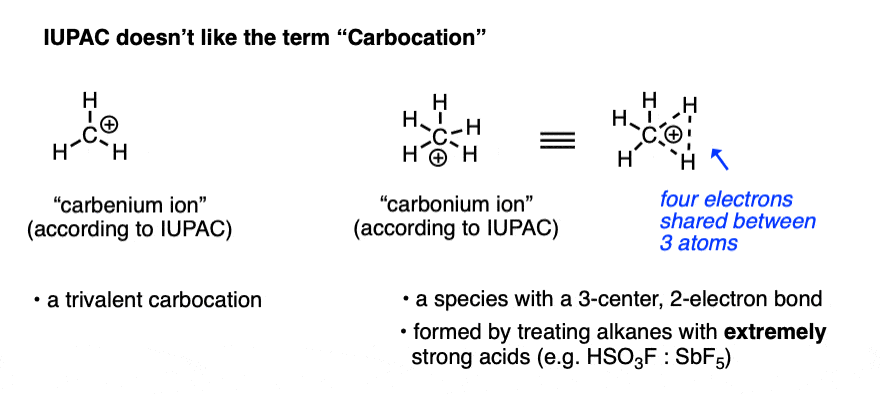
Note 1. For most purposes, “carbocation” refers to a carbon with three bonds and an empty orbital, and in the vast majority of cases, this is the terminology that is used. There is a lot of inconsistency in the older literature as to whether these are “carbonium” or “carbenium” ions. The isolation of the CH5(+) species in extremely strong acid solution by the research group of George Olah at USC led IUPAC to define the term “carbenium ion” as a trivalent carbon and “carbonium ion” as a five-coordinate carbon. [Ref].
Note 2. Two prominent exceptions to this are the vinyl carbocation and the acylium ion.
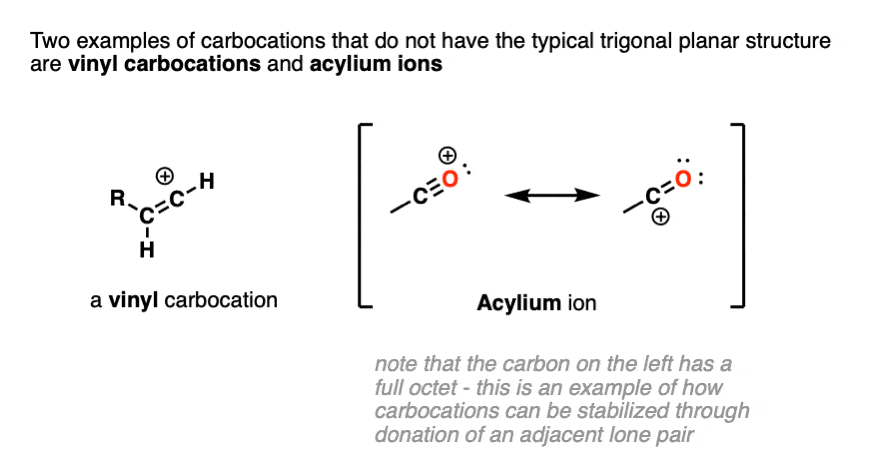
Note that the vinyl carbocation has a linear geometry and the central carbon is sp-hybridized.
The acylium ion is an excellent example of a carbocation that is stabilized through donation of an adjacent lone pair. (For examples of the acylium ion, see – The Friedel Crafts Alkylation and Acylation).
Note 3. The structures of several carbocations have been determined through X-ray crystallography.
For example, the structure of the t-butyl cation shows a trigonal planar structure (bond angles 120°) with C-C bond length of 1.44 Angstroms [Ref] which is about halfway between a typical C-C single bond (1.50 Å) and a C-C double bond (1.40 Å)
The structure of the adamantyl cation is particularly interesting. In adamantane, all C-C bonds are approximately the same length (1.53 Å). However, in the adamantyl carbocation, the carbon directly bonded to the carbocation becomes noticably shorter (1.43 Å) while the C-C bond that is able to overlap with the p-orbital of the carbocation is noticeably longer (1.608 Å).
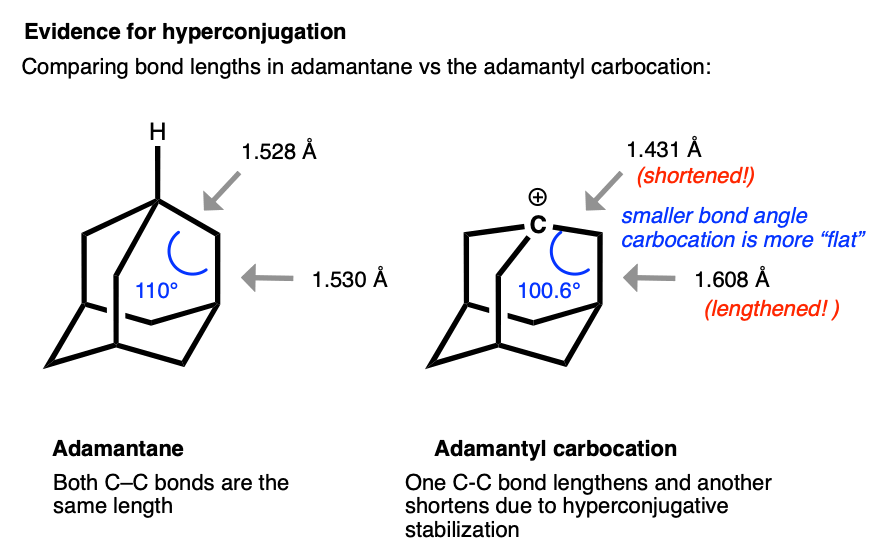
This is exactly what we would have predicted from using the hyperconjugation model for carbocation stabilization, where one C-C bond gains partial double bond character and another bond is weakened.
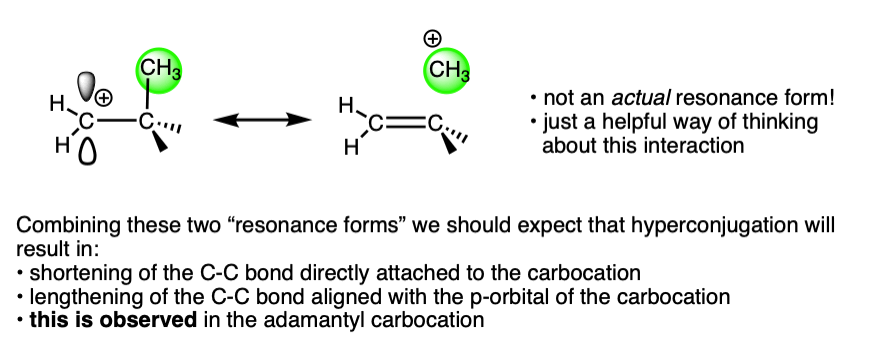
For a lot more detail on this, I highly recommend lecture 30 from Prof. David Evans’ Advanced Organic Chemistry 206 at Harvard, link here.
Note 4. One way to quantify the stability of carbocations is through measuring their hydride affinities, that is, the energy released upon reaction of the carbocation with a hydride ion. The hydride affinities of a large number of carbocations have been measured in the gas phase (where they have longer lifetimes).
A selection is provided below:
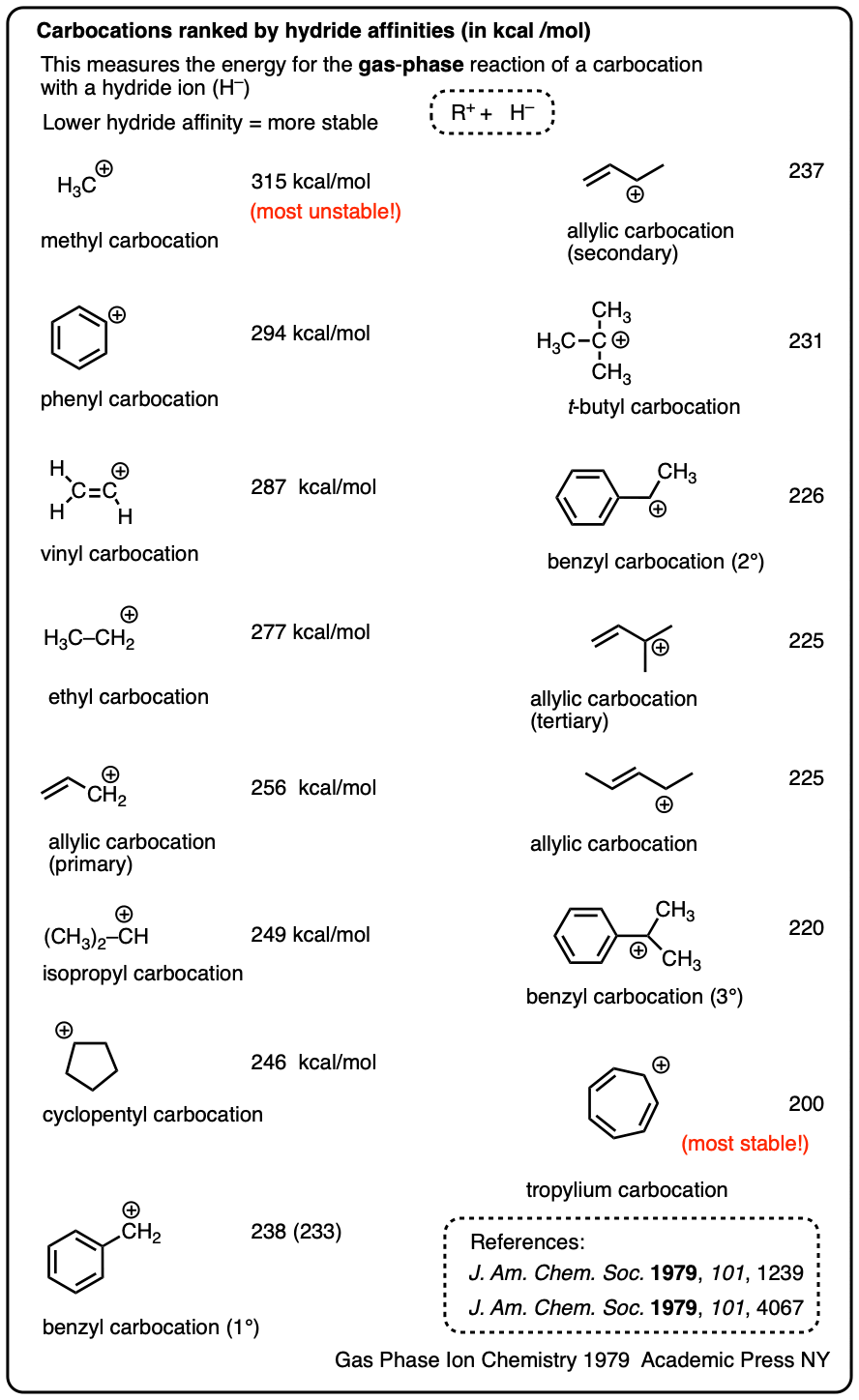
Solution-phase hydride affinities have also been measured for some of the more stable carbocations (gas phase hydride affinities in parentheses)
- Ph3C(+) : 96 kcal/mol
- Ph2CH(+): 105 kcal/mol
- PhCH2(+): 118 kcal/mol (238 kcal/mol)
- Tropylium ion: 83 kcal/mol (200 kcal/mol)
Note 5. Carbocation formation is easier in solvents with a high dielectric constant such as water or carboxylic acids (e.g. acetic acid, formic acid).
Note 6 One class of tertiary carbocations that are particularly unstable are carbocations that are constrained in a bridgehead. Ring strain prevents these carbocations from adopting a trigonal planar geometry.
Note 7. Carbocations that are part of small rings such as cyclopropyl and cyclobutyl carbocations are also unusually unstable.
Note 8. An alternative method of measuring carbocation stability is through measuring rates of hydrolysis in a nucleophilic solvent. However, as this graphic from Evans shows, these two measures do not always correlate well…

Quiz Yourself!
 Click to Flip
Click to Flip
 Click to Flip
Click to Flip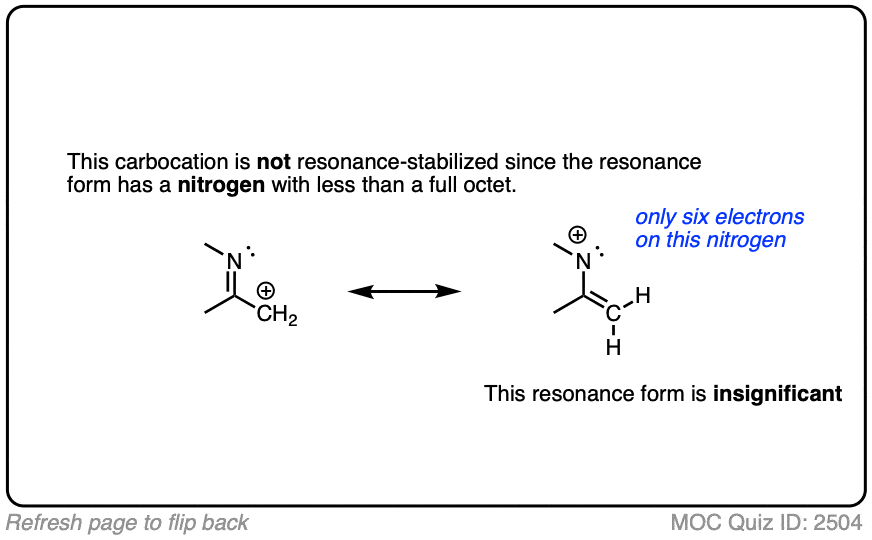
 Click to Flip
Click to Flip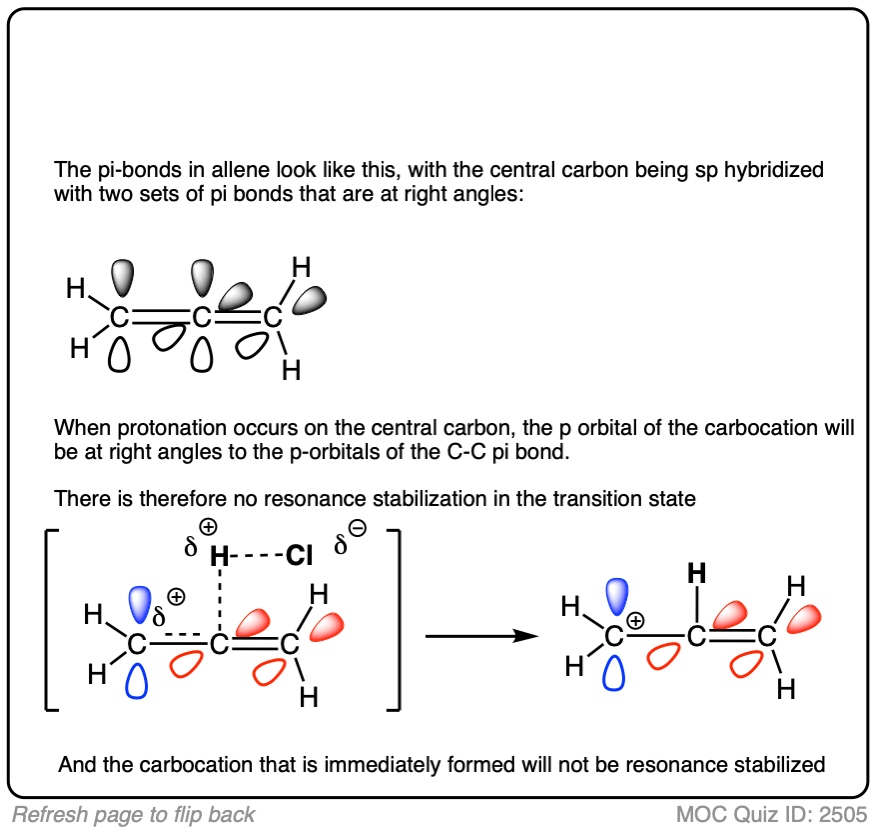
 Click to Flip
Click to Flip